“嘿,Siri,明天广州下雨吗?”
“明天广州应该不会下雨。”
图1. 与Siri 对话界面。
以上情景经常出现在我们的生活中,实现和语言助手进行人机交互的一个重要技术是语音识别,而这正属于人工智能(Artificial Intelligence, AI)的一种应用。
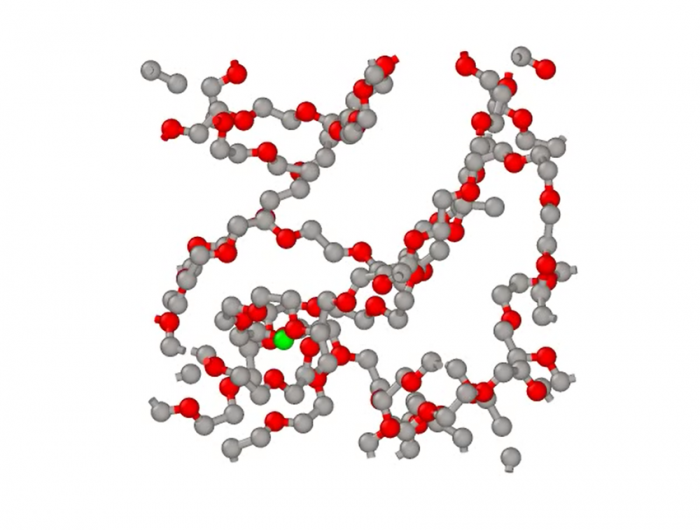
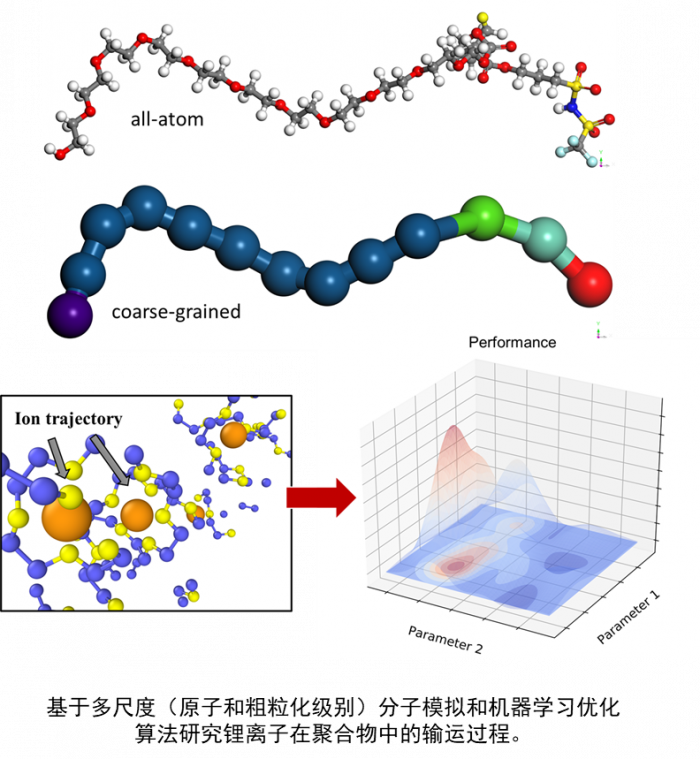
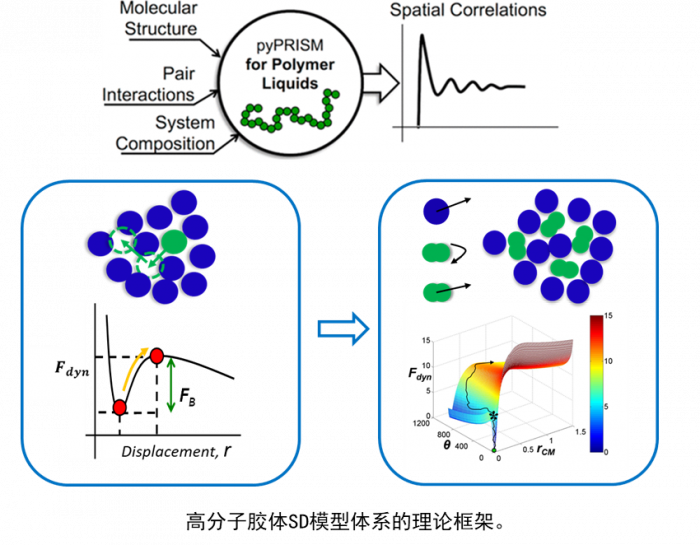
大数据和人工智能的结合被称为“第四科学范式”[1]。机器学习方法作为人工智能重要的支柱之一,近年来受到了广泛的关注。在材料科学领域,由于数据的丰富和计算机运算能力的增强,机器学习方法已经被应用于发现新材料、预测材料和分子性质、研究原子力场和设计药物等多个方向。
接下来我们将简单介绍机器学习方法的概念、分类、基本步骤和常用的软件库。
机器学习方法的概念
对于机器学习,Mitchell给出了一个形式化的定义:一个计算机程序在完成任务T之后,获得经验E,其表现效果为P,如果任务T的性能表现,也就是用以衡量的P,随着E的增加而增加,可以称其为学习[2]。
这种通过计算的方式,利用数据来改善系统自身性能的能力在材料科学领域的应用数量正在迅速增加。用于生成、测试和改善科学模型的机器学习工具也越来越多。这些技术适用于解决设计大量组合空间或非线性过程的复杂问题,而常规过程无法解决这些问题或需要很高的计算成本。运用机器学习,只要提供足够的数据和用于发现规则的算法,计算机原则上就有能力在没有人类干预的情况下确立所有已知的物理规律,以及可能发现目前还未知的物理规律[3]。
机器学习方法的分类
机器学习方法按学习方式划分大体可以分为三大类,分别是监督学习、无监督学习和强化学习[4],见图2。
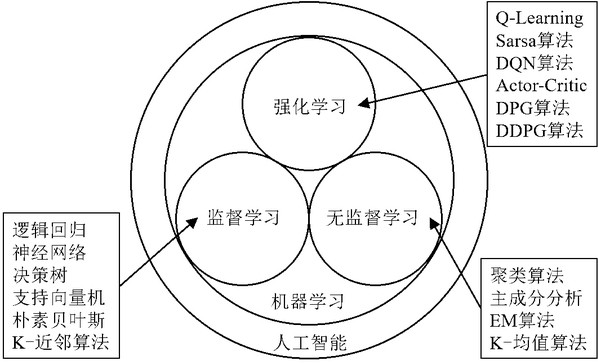
图2. 机器学习方法分类。
监督学习的特征是训练数据有标记信息,训练数据由输入和对应输出值的集合组成。两种常见的监督学习问题是分类和回归,对应于分类输出和数值输出。常见的方法有线性回归、逻辑回归、支持向量机和神经网络等。
无监督学习的特征是训练数据无标记信息,尝试从没有标记的数据中提取信息。无监督学习分为非概率模型,如稀疏编码、自动编码器、k-means等,以及概率(生成)模型,其中或明确地或隐含地涉及密度函数。常见的方法有聚类、降维以及异常检测等。
强化学习是关于一个智能体与环境相互作用,通过试错来学习最优策略,用于自然、社会科学和工程等广泛领域的序贯决策问题[5]。在强化学习中,有评估反馈,但没有监督信号。
机器学习方法的基本步骤
在传统的计算方法中,计算机更多情况是充当高级计算器的角色,所有步骤都已经由人提供了硬编码算法,即将数据直接写在程序或其他可执行对象的源代码中。与传统计算方法不同,机器学习方法通过评估数据集的一部分并建立模型进行预测来学习数据集的基础规则。计算机能通过数据的内部联系发现规律,所以我们说机器从数据中进行了“学习”。
在材料科学中,训练机器学习模型的四个步骤主要可以分为数据采集、数据表达、模型选择和模型优化[6]。
数据采集

图3. 数据库标识。(1)无机晶体结构数据库;(2)剑桥结构数据库。
这些数据虽然已经经过一定的筛选和检查,但是难免会受到人为和测量误差的影响。此外,由于数据以各种格式存储在不同的数据库中,因此很难将多个数据库的数据统合起来。所以,即使使用数据库中已经收集的数据用于机器学习模型的训练,也应该在数据采集阶段进行相应的处理。
数据表达
数据表达又可称为特征工程,是指将原始数据转换或提取信息特征以适应算法学习的形式。不同的数据形式对于机器学习算法的训练影响巨大。如何最好地表达数据是一个值得思考的问题,它不仅仅涉及研究者对于研究体系的认知,还涉及机器学习算法对数据的处理过程。图4是分子表示(Representation of molecules)的示意图。
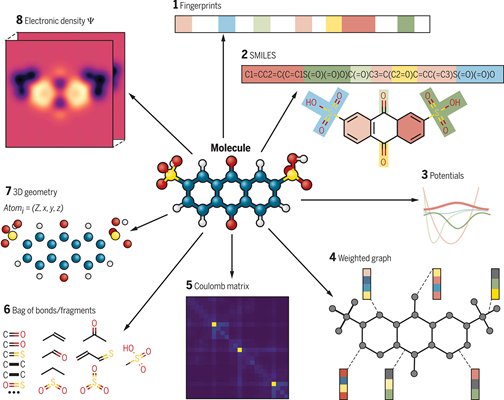
图4. 对于同一个分子不同类型的分子表示[7]。(1)指纹矢量;(2)SMILES字符串;(3)势能函数;(4)原子和键的权重图;(5)库伦矩阵;(6)键/片段的组合;(7)原子电荷的三维几何;(8)电子密度。
有时最方便人类理解问题的表达形式并非适合机器对其进行“学习”。例如在固态下,传统的使用平移矢量和原子的分数坐标来描述晶体结构的方法并不适用于机器学习,因为晶格可以通过选择不同的坐标系从而以无数种方式来表示。利用基于径向分布函数的表示法是解决这一问题的新方法之一[8]。
模型选择
完成数据采集和数据表达后,已经有了足够的数据以及合适的数据表达,这一步则是在前两步的基础上建立一个用于学习的模型。根据需要解决的问题以及可用数据的类型和数量,机器学习模型的训练可以采用监督、无监督和强化学习方法。
监督学习是目前最成熟的方法,通过输出值对预测值的校正通常能获得比较好的学习模型。无监督学习可用于更全面的数据分析和分类,或识别大型数据集中以前无法识别的模式。强化学习没有监督信号,只有奖励(反馈)信号。该方法不需要事先给出任何数据,而是通过接收环境对动作的奖励(反馈)来获取学习信息并对模型参数进行更新。
每种算法都有自己适用的领域,并且没有针对所有问题都通用的最佳算法,这就是“没有免费的午餐”定理(No free lunch theorem)。研究人员应该根据数据集的构成和研究目的,选择适当的机器学习方法。
模型优化
为了优化和选择模型的最佳模式,模型必须进行验证评估。我们通常把数据分成训练集、验证集和测试集三大部分。判断已建立的机器学习模型表现好坏通常需要用验证集数据进行评估,即通过验证集数据对模型的泛化误差进行评估。
两个常见的问题是过拟合和欠拟合,前者体现了机器学习模型的学习能力较差,难以达到所需要的预测或分类等效果,后者体现了机器学习模型的泛化能力较差,对于训练集的数据能够较好地拟合但是对于训练集之外的数据拟合能力很差。通过优化模型和数据集,平衡这两个问题的行为就是偏差-方差权衡(Bias-variance trade-off)。
机器学习方法常用的软件库
机器学习方法多种多样,幸运的是,有很多成熟的库可供我们直接使用,不需要重新“造轮子”。常用的库有Scikit-Learn、Keras以及TensorFlow,见图5。

图5. 三种常见的机器学习库。(1)Scikit-Learn;(2)Keras;(3)TensorFlow。
Scikit-Learn用于Python编程语言的自由软件机器学习库。它的特征是具有各种分类、回归和聚类算法,包括支持向量机、随机森林、梯度提升、k-平均聚类和DBSCAN。
Keras是一个用Python编写的开源神经网络库,旨在快速实现深度神经网络,专注于用户友好、模块化和可扩展性。除标准神经网络外,Keras还支持卷积神经网络和循环神经网络。
TensorFlow是一个用于机器学习的免费开源软件库。它可以用于一系列任务,但特别着重于深度神经网络的训练和推理。
通过使用适合的机器学习库,即使对于机器学习底层算法不熟悉的同学,也可以轻松上手机器学习,搭建属于自己的机器学习模型。
参考文献
[1] Agrawal A, Choudhary A. Perspective: Materials informatics and big data: Realization of the fourth paradigm of science in materials science[J]. Apl Materials, 2016, 4(5): 053208.
[2] Mitchell T M. Machine learning[J]. Burr Ridge, IL: McGraw Hill, 1997, 45(37): 870-877.
[3] Butler K T, Davies D W, Cartwright H, et al. Machine learning for molecular and materials science[J]. Nature, 2018, 559(7715): 547-555.
[4] Li Y X. Deep reinforcement learning: An overview[J]. arXiv preprint arXiv, 2017, 1701.07274.
[5] Sutton R S, Barto A G. Reinforcement learning: An introduction[M]. MIT press, 2018.
[6] Butler K T, Davies D W, Cartwright H, et al. Machine learning for molecular and materials science[J]. Nature, 2018, 559(7715): 547-555.
[7] Benjamin S L, Alán A G. Inverse molecular design using machine learning: Generative models for matter engineering[J]. Science, 2018, 361: 360-365.
[8] Schütt K T, Glawe H, Brockherde F, et al. How to represent crystal structures for machine learning: Towards fast prediction of electronic properties[J]. Physical Review B, 2014, 89(20): 205118.